Sistema inmune, microinflamación y permeabilidad
En este apartado haremos un viaje por algunas de las alteraciones inmunitarias encontradas en este síndrome, relacionándolas con síntomas como el dolor y la dismotilidad, o con otros hallazgos frecuentes como el aumento en la permeabilidad intestinal. A día de hoy esta es una de las líneas de investigación biológica con más trayectoria en SII, y algunos conceptos, como la activación mastocitaria, tienen ya más de 30 años de recorrido. El SII es más frecuente en personas con problemas inmunitarios como asma, alergias, dermatitis atópica o artritis reumatoide (ver 3.1.3), por lo que no debería extrañarnos que el sistema inmune esté implicado en su patofisiología.
¿INFLAMACIÓN O MICROINFLAMACIÓN?
En el SII parecen existir procesos micro-inflamatorios distintos de la inflamación habitual que aparece en enfermedades como las inflamatorias intestinales (Crohn y colitis ulcerosa), la cual es visible con radiografías y técnicas endoscópicas. En el SII esta inflamación es invisible por estos métodos (se necesitan biopsias y estudios microscópicos de mediadores -más que células- inflamatorios), y no puede ser medida con las pruebas de inflamación habituales como la proteína C reactiva (CRP en inglés), o la calprotectina en heces. De estas dos pruebas, la calprotectina es específica del tracto digestivo (se mide a través de las heces), y la CRP es más general, suele usarse como marcador de riesgos para problemas cardiovasculares (y se mide a través de la sangre). Estas pruebas son, pese a todo, una buena forma de discriminar entre un SII o una EII, y los digestivos, cuando tienen dudas, suelen mandarlas antes de procedimientos más agresivos como la colonoscopia. Según un metaanálisis de 2015, valores inferiores a 40 en la calprotectina fecal y a 0.5 en la proteína C reactiva reflejan una probabilidad inferior al 1% de tener una EII, lo que orientaría el diagnóstico hacia condiciones diferentes como el SII. Hay estudios que muestran pequeñas variaciones en los valores de calprotectina y CRP en pacientes con SII, pero sin llegar a los niveles de las EII.
La micro-inflamación del SII podría estar relacionada, como veremos después, con el nervio vago, cuya función antiinflamatoria está alterada en estos pacientes. El SNA parasimpático, al que este nervio pertenece, se sirve de la acetilcolina (neurotransmisor) y una serie de enzimas hidrolíticas (enzimas que degradan por hidrólisis, es decir, agregando una molécula de agua) para cumplir su función antiinflamatoria. Un aumento en la actividad de la colinesterasa sérica (o pseudocolinesterasa), enzima que hidroliza la acetilcolina, puede disminuir la inhibición de la inflamación (y por tanto, provocar inflamación). En un estudio de 2018 se observó un incremento significativo de la colinesterasa sérica en pacientes de SII-D en comparación con sujetos sanos. Este aumento en la colinesterasa sérica se asoció a valores significativamente mayores de inflamación, medidos por la proteína C reactiva, que pese a todo se mantuvo en el rango de la normalidad que antes comentamos.
No obstante, cuando nos referimos a que en el SII, al menos en ciertos subtipos, puede existir una inflamación crónica de bajo grado o micro-inflamación, no hablamos de la que se mide con CRP o calprotectina, sino de procesos intracelulares muy trabajosos de medir, y que no siempre se asocian a los síntomas. Para entender mejor estos procesos, primero hablaremos de la permeabilidad intestinal.
PERMEABILIDAD PARACELULAR
En el apartado de alteraciones de la microbiota, cuando hablamos de los productos de la fermentación, vimos cómo varios procesos pueden afectar a y verse afectados por la permeabilidad intestinal. La barrera intestinal (que se detalla también en dicho apartado) cuenta con varias capas, pero por ahora vamos a hablar del transporte paracelular. Éste hace referencia al transporte de sustancias a través del epitelio pasando por el espacio intercelular o «entre células», en contraposición al transporte transcelular, en el que las sustancias tienen que atravesar la célula.
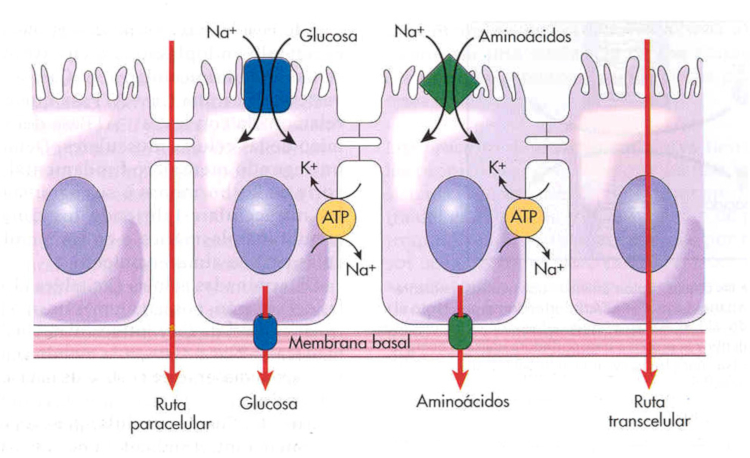
La permeabilidad paracelular es la facilidad con la que los contenidos del lumen (patógenos, toxinas, bacterias, nutrientes, ácidos grasos de cadena corta, productos varios de la fermentación…) atraviesan la unión entre células y penetran en los tejidos. La célula regula este canal paracelular mediante proteínas de unión estrecha, que reducen la permeabilidad y crean un “sistema de filtrado” más eficiente.
Una revisión de 2014 comienza mencionando un estudio en el que se encontró que, en la mucosa colónica de pacientes con SII-diarrea o SII-mixto, puede haber una disminución de E-cadherinas, un tipo de proteínas de unión estrecha que regulan esta permeabilidad paracelular (otro estudio de 2011 habla de un disminución significativa de otras dos proteínas de unión estrecha, la ocludina y la claudina-1, sólo en el subtipo SII-diarreas). En la misma revisión se habla de un incremento en el número de células inmunitarias, como los mastocitos o los linfocitos T, en algunos subgrupos de pacientes con SII. También podría haber un aumento en el número de Células EnteroEndocrinas (CEE, EEC en inglés, son células neuroendocrinas que se ubican junto a las células epiteliales del intestino, aunque principalmente están en el estómago). Es precisamente a estas células, y a su actividad, a lo que nos referimos cuando hablamos de micro-inflamación. En el resumen del artículo se dice textualmente:
“los mediadores inflamatorios liberados por estas células u otros factores luminales podrían estar en el origen de las funciones alteradas de la barrera epitelial y de la señalización del sistema nervioso entérico, que llevan a la hipersensibilidad visceral”
A día de hoy, esto es sólo una teoría, y no se puede descartar que ocurra al revés (como planteaba Quigley en la foto de arriba del eje microbiota-intestino-cerebro), y sea el aumento de permeabilidad el causante de esta micro-inflamación, por filtración de partículas que las proteínas de unión estrecha no dejarían pasar de otro modo, generándose así la respuesta inmune.
En uno de los estudios sobre permeabilidad intestinal más interesantes realizados hasta la fecha, los investigadores tomaron sobrenadantes (partículas que flotan al meter la biopsia en un recipiente con agua) de biopsias de pacientes con SII, y sobrenadantes de biopsias de personas sanas. Después tomaron varios modelos in vitro de una barrera intestinal humana, para lo que usaron células Caco-2. Las células Caco-2 son células derivadas de carcinomas del colon de seres humanos, pero, cuando se realiza un cultivo con ellas bajo unas condiciones determinadas, estas células sufren una diferenciación que les lleva a imitar, tanto en su morfología como en su funcionamiento, a los enterocitos o células epiteliales del intestino delgado (presentan proteínas de unión estrecha, microvellosidades, y enzimas y transportadores específicos de estas células). Los investigadores en este estudio expusieron estos modelos in vitro Caco-2 a los sobrenadantes de pacientes de SII (9 SII-D, 8 SII-C, 11 SII-M), y vieron si había cambios en la permeabilidad paracelular. Se encontró que los modelos expuestos a sobrenadantes de pacientes con SII aumentaron su permeabilidad significativamente, sin importar el tipo de SII, y este aumento en la permeabilidad correlacionó significativamente con la severidad y frecuencia del dolor y la distensión abdominal. Aquí el estudio de 2018. En él también se habla de los efectos de un probiótico capaz de revertir ese aumento de permeabilidad (Escherichia coli Nissle 1917), pero es muy probable que los efectos en un modelo in vitro no se repitan en el intestino humano, pues el efecto terapéutico se acabaría diluyendo en la interacción de esta cepa con trillones de bacterias diferentes, y el Mutaflor (el probiótico que contiene esta cepa) no ha mostrado una efectividad clara en los pocos estudios controlados que se han hecho en SII.
Otro estudio muy interesante, de 2014, utilizó endomicroscopía láser confocal (una técnica endoscópica que permite hacer estudios histopatológicos – estudios microscópicos de los tejidos – en tiempo real) y biopsias para ver los cambios en las células epiteliales del duodeno en pacientes con SII a los que se les introducían diferentes antígenos alimentarios (leche de vaca, trigo, levadura y soja). Se observó que, en 22 de 36 pacientes, la mucosa duodenal se alteró inmediatamente después de introducir alguno de los antígenos. En estos 22 pacientes que respondieron, el número de linfocitos intraepiteliales que tenían de base (antes de la prueba) ya era significativamente mayor que en los otros 14 pacientes. Además, en estos 22 pacientes, 5 minutos después de la exposición a estos antígenos, los linfocitos intraepiteliales aumentaron, aunque de manera no significativa, y aparecieron separaciones o «huecos» entre las células epiteliales, y espacios aumentados entre las microvellosidades, siendo ambos aumentos significativos (aumento de la permeabilidad). Los 22 pacientes que mostraron alteraciones duodenales mejoraron con una dieta de exclusión de los antígenos a los que respondieron (hasta un 74% a los 12 meses), aunque no se contó con un grupo control para esta aseveración (a los otros 14 no se les dio ninguna dieta).
En 2021, se descubrió que, al inyectar ciertos antígenos alimentarios (gluten, trigo, soja, leche) en la mucosa rectosigmoidea de pacientes con SII, se generaba edema en la zona y activación mastocitaria, aunque no se aclaró si estos antígenos provocaban síntomas o no.
También se ha observado que el aumento de la permeabilidad colónica en pacientes con SII se asocia a un aumento en el paso de bacterias comensales y patógenas a través de la membrana epitelial. Un estudio de 2017 lo demostró utilizando microscopía electrónica y biopsias colónicas en cámaras de Ussing, observándose que las bacterias Escherichia coli HS y Salmonella typhimurium (marcadas con fluorescencia) cruzaban el epitelio colónico en mayor número en biopsias de SII, y que además sólo lo hacían por ruta transcelular (a través de la célula) en vez de paracelular (por las uniones estrechas entre células epiteliales). Los anticuerpos de VPACs (receptores del péptido intestinal vasoactivo o VIP) y el ketotifeno (estabilizador de mastocitos y antihistamínico) reducían este paso de bacterias en modelos in vitro. Además, en pacientes SII había un aumento en los niveles de triptasa (enzima proteolítica con función proinflamatoria y segregada por los mastocitos), en el número de mastocitos, y en el número de mastocitos que expresaban el receptor VPAC1. Todo esto parece confirmar que tanto el VIP como los mastocitos son mediadores de este problema de permeabilidad. El estudio también mostró que las bacterias como la Salmonella podrían tener efectos perniciosos sobre la permeabilidad intestinal una vez que han penetrado en la célula, pues cuando añadieron esta bacteria a biopsias de SII se redujeron significativamente los niveles de ocludina (proteína de unión estrecha), pero este efecto se revirtió considerablemente al añadir también ketotifeno.
ANTICUERPOS (INMUNOGLOBULINAS) Y RESPUESTA INMUNE
Los anticuerpos (o inmunoglobulinas) son proteínas que se encargan de identificar y neutralizar antígenos potencialmente tóxicos como determinadas bacterias o virus (1993). Son sintetizados por los linfocitos B. Su estructura es bastante similar, pero una región del ápice de la proteína es extremadamente variable (región hipervariable), permitiendo la existencia de millones de anticuerpos. Cada una de estas variantes se une a una «diana» distinta, que es lo que conocemos como antígenos (2001), a través del epítopo (el área del antígeno que es reconocida por el anticuerpo). La función de los anticuerpos es «marcar» a los antígenos, para que otras células del sistema inmunitario los reconozcan y eliminen, en un proceso al que llamamos «respuesta inmune».
En mamíferos, existen 5 isotipos o clases de anticuerpos humorales:
- IgA: Presente en las mucosas, como el tracto digestivo, respiratorio y urogenital, impidiendo su colonización por patógenos como bacterias o virus (también están en la saliva, la sangre y la leche). Hay 2 subtipos (1986) y representan el 13% de las Ig.
- IgD: Actúa como receptor de antígenos en los linfocitos B que aún no han sido expuestos a éstos, y desaparece tras ocurrir dicha exposición (2006). Son el 0.2% de las Ig.
- IgE: Se une a alérgenos y desencadena la liberación de histamina y citocinas proinflamatorias desde los mastocitos, eosinófilos y basófilos. Protege principalmente contra parásitos helmintos (2004). Son menos del 0.2% de las Ig.
- IgM: Se expresa en la superficie de los linfocitos B y es la primera Ig que se sintetiza en respuesta a una infección (respuesta primaria, cuando un antígeno nos infecta por vez primera), hasta que la respuesta inmune humoral avanza y se sintetizan suficientes IgG (2006). Son el 6% de las Ig.
- IgG: Es la Ig predominante en los fluidos internos del cuerpo, y la única que atraviesa la placenta para dar inmunidad pasiva al feto (2004). Se produce como respuesta a la invasión por bacterias, hongos o virus, y está implicada principalmente en la respuesta secundaria (cuando un antígeno que nos ha infectado antes intenta invadir por segunda vez). Hay 4 subtipos y son más del 80% de las Ig.
Los anticuerpos (Ig) se unen a los antígenos por el epítopo en un mecanismo llamado reacción antígeno-anticuerpo, que forma un complejo del mismo nombre. La unión es específica (un anticuerpo para un antígeno), reversible, rápida y espontánea (no conlleva gasto de energía). Esta unión puede darse por precipitación (los anticuerpos se unen a antígenos con varios epítopos, y éstos dejan de ser solubles, siendo más fácil su fagocitación), aglutinación (si hay varias células infectadas y los anticuerpos se unen a ellas, a veces se forman agregados de éstas que, nuevamente, serán más fáciles de fagocitar), neutralización (virus y toxinas que quedan anuladas cuando el anticuerpo se les une) y opsonización (cuando los anticuerpos se unen al antígeno, éste es más reconocible para los macrófagos y neutrófilos que los fagocitan).
Cuando un antígeno entra por primera vez en el organismo, existe lo que se llama respuesta primaria, que consta de 3 fases:
- Fase de latencia: Tenemos los síntomas de la enfermedad y el organismo produce linfocitos que reconocerán al antígeno y producirán anticuerpos.
- Fase logarítmica: El nivel de anticuerpos IgM en sangre se dispara.
- Fase de declinación: Conforme se erradica la infección, los IgM van descendiendo, pero se mantienen algunas células como memoria (células B de memoria).
Si ese mismo antígeno vuelve a intentar entrar, ocurre la respuesta secundaria, con 2 fases:
- Fase de latencia: Más breve que la anterior, ni notamos síntomas.
- Producción de anticuerpos: Mucho más rápida que la vez anterior (las células de memoria recuerdan al antígeno), en mucha mayor cantidad, y esta vez son del tipo IgG.
Este proceso, así como el rol de algunas células del sistema inmune, puede entenderse mejor si explicamos antes las dos respuestas inmunitarias principales de nuestro organismo ante una infección: la humoral (de los humores, el plasma intersticial, el ambiente extracelular) y la celular (del interior de la célula).
La respuesta inmunitaria HUMORAL tiene como protagonistas a los anticuerpos, así como a las células que los producen (linfocitos B), y ataca a patógenos extracelulares (que se mueven entre células) y sus toxinas, o a patógenos intracelulares que salen al medio extracelular para contagiar a otra célula. Para entender esta repuesta tenemos que conocer la teoría de la selección clonal, según la cual, desde antes de la infección ya existirían en nuestro cuerpo linfocitos B que producen anticuerpos para todos los antígenos que existen (generados por las células madre durante el desarrollo embrionario). Como resultado, tendríamos linfocitos B con anticuerpos específicos para antígenos que aún no han llegado al cuerpo. Cuando finalmente llegan los antígenos, se unirían al linfocito B que tuviera anticuerpos específicos contra ellos en su superficie, el cual aumentaría de tamaño y proliferaría rápidamente, dando lugar a numerosos clones (selección clonal) que se diferenciarían en dos subtipos:
- Células B plasmáticas: Más grandes, con un citoplasma compuesto casi en su totalidad por un retículo endoplasmático rugoso que sintetiza anticuerpos IgM.
- Células B de memoria: Implicadas en la respuesta secundaria, se mantienen en el organismo y, en caso de una segunda infección por el mismo antígeno, se encargan de dar lugar a células B de ambos subtipos, aunque en vez de IgM producirán IgG. Al proceso por el que los linfocitos B plasmáticos dejan de producir un tipo de Ig para producir otra se le conoce como «conmutación isotípica». Normalmente los linfocitos B vírgenes (que aún no han conocido antígeno) producen IgM e IgD, y, cuando son activados por el antígeno y se convierten en linfocitos «maduros», proliferan y comienzan a producir altos niveles de anticuerpos. Sin embargo, si estos linfocitos B son activados por obra y gracia de los linfocitos Th (o T colaboradores), como ocurre en la respuesta inmunitaria celular que ahora veremos, tiene lugar la conmutación isotípica y pasan a producir IgG, IgA o IgE (2004).
En la proliferación y diferenciación de linfocitos B tienen un importante rol mediadores como el «factor activador de células B» o BAFF (2011), una citocina que pertenece a la familia TNF y que se expresa en células de linaje de células B. Las células B «maduras» tienen receptores específicos para esta citocina (BAFF-R, TACI, BCMA).
La respuesta inmunitaria CELULAR es la mediada por los linfocitos T y otros agentes (macrófagos, neutrófilos…) contra patógenos intracelulares (en el interior de la célula) o tumorales. Aquí tiene un rol clave el complejo mayor de histocompatibilidad (MHC en inglés), que se encarga de presentar el antígeno a las células del sistema inmunitario, y del que hay dos subtipos. El MHC-I esta en todas las células, y funciona cuando éstas se vuelven tumorales o son infectadas, en cuyo caso es reconocido por los linfocitos T citotóxicos (Tc) CD8. El MHC-II está en unas células del sistema inmune llamadas células presentadoras de antígeno (APC), que presentan a éste unido al MHC-II y son reconocidas por los linfocitos T colaboradores (Th) CD4. La respuesta celular consta de varios pasos:
- Los macrófagos fagocitan los antígenos, los digieren en su citoplasma y los fragmentos del antígeno suben a la superficie de la célula, donde se unen al MHC-II, transformándose así el macrófago en una APC. Los linfocitos Th CD4 reconocen a la APC y, a través de mensajeros químicos, activan a los linfocitos B (que inician la respuesta humoral específica para ese antígeno), a los macrófagos y otros fagocitos (para que fagociten más) y a los linfocitos Tc CD8.
- Estos Tc CD8 se activan también al contactar con la célula APC, y su función es destruirla, mediante citotoxinas (degradan la membrana celular), citocinas (impiden la replicación de virus) o linfocinas (activan a los macrófagos).
- En paralelo, las células B y T de memoria se desarrollarán para preparar la respuesta secundaria en caso de reinfección, las células NK (Natural Killer) matan a células infectadas de manera inespecífica, y los linfocitos T supresores inhiben a los Th y frenan la respuesta humoral y celular cuando ya se ha erradicado la infección.
Los mensajeros químicos utilizados durante el proceso son, entre otros, los interferones (IFN, interfieren en la replicación vírica), los factores de necrosis tumoral (TNF, citocinas que matan a células cancerígenas), o las interleucinas (IL, citocinas que activan o inhiben a otros leucocitos del sistema inmune).
Pero, ¿qué relación tiene todo esto con el SII?
Un estudio de 2020 investigó el rol de los anticuerpos IgA ileales (del íleon) y de las bacterias revestidas de anticuerpos IgA en el SII-D. El estudio utilizó biopsias ileales y cecales (del ciego), muestras de heces y analíticas de sangre. En las muestras de heces, se encontraron niveles más elevados de IgA-1 en los pacientes SII-D. También encontraron que la conmutación isotípica a IgA y los receptores de BAFF (BAFF-R) estaban aumentados en el íleon terminal de pacientes SII-D. La composición de la microbiota también variaba entre pacientes y controles, y se observó que los pacientes presentaban una mayor proporción de bacterias revestidas por IgA (bacterias IgA+). Las bacterias IgA+ de los SII-D tenían mayor abundancia de Escherichia-Shigella, Granulicatella, y Haemophilus en comparación con las bacterias IgA+controles y con las IgA- (bacterias no revestidas por IgA) de los propios SII-D. Los niveles de IgA recubriendo bacterias Escherichia-Shigella correlacionaban positivamente con ansiedad y depresión. Por otro lado, la proporción total de bacterias Escherichia-Shigella, la actividad IgA luminal y algunas bacterias alteradas IgA+ correlacionaban positivamente con los síntomas del SII-D (alteraciones de la motilidad y dolor abdominal visceral). Los autores concluyeron que la disbiosis microbiana podría promover que la mucosa ileal terminal produzca niveles más altos de IgA, aumentando la proporción de bacterias recubiertas de IgA (IgA+) al activar la conmutación isotípica de esta inmunoglobulina, lo que podría regular la inflamación local y las manifestaciones clínicas en el SII-D. En resumen, la IgA podría mediar los efectos de la disbiosis microbiana en la patogénesis del SII-D.
CITOCINAS Y LINFOCITOS
Ahora que tenemos unas nociones básicas sobre la permeabilidad intestinal, volvemos al tema de la microinflamación. Este breve vídeo, con un recordatorio básico de los tipos de células de nuestro sistema inmune, nos ayudará a entender los párrafos sucesivos:
En varios estudios con pacientes de SII se habla de un aumento de células inmunitarias refiriéndose a los mastocitos, monocitos (un tipo de glóbulo blanco) y macrófagos (el estadio posterior a los monocitos, cuando éstos, tras X horas, dejan de circular por el torrente sanguíneo y se fijan a un tejido determinado). Respecto al rol de las citoquinas (o citocinas, un tipo de proteína del sistema inmune) en el SII, lo primero es aclarar que éstas pueden ser proinflamatorias o antiinflamatorias, según su efecto sobre otras células inmunitarias. Hay varios estudios hasta la fecha:
- Un estudio (2018) encontró que los niveles plasmáticos (en sangre) de citocinas proinflamatorias, incluyendo entre éstas el factor alfa de necrosis tumoral (TNF-α) o las interleucinas IL-6, IL-8 o IL-1β, estaban aumentados en algunos subgrupos de SII, aunque no parecían relacionarse con los síntomas.
- Otro estudio (2017) observó un aumento de las citoquinas proinflamatorias (TNF-α, IL-17) plasmáticas en pacientes con SII, y un descenso de las antiinflamatorias (IL-10) en los SII-D, que en este caso sí se relacionaban con la sintomatología.
- Esta correlación se repitió en un estudio de 2013, donde las citoquinas esta vez se obtuvieron de los sobrenadantes de biopsias de distintos pacientes (permitiendo ver la concentración de estas citocinas a nivel local, en el colon), y la frecuencia e intensidad del dolor volvieron a relacionarse con la elevación de las citoquinas IL-1β y TNF-α, ambas proinflamatorias.
- Otro estudio, de 2017, midió nuevamente los niveles plasmáticos de citocinas proinflamatorias y antiinflamatorias en pacientes con SII, llegando una vez más al mismo resultado, las proinflamatorias (en este caso, TNF-α, IL-17 y MDA) estaban aumentadas y las antiinflamatorias (IL-10) reducidas, y todas correlacionaban con los síntomas.
- Un metaanálisis previo (2014) ya había obtenido resultados parecidos, encontrando niveles plasmáticos de TNF-α superiores a los normales en pacientes SII de subtipos específicos y en pacientes mujeres, así como niveles plasmáticos de IL-10 inferiores a los normales en pacientes varones. En el estudio también analizaron biopsias de colon (concretamente, los niveles de ARNm, implicado en la síntesis de proteínas), encontrando una expresión reducida de IL-10 en el colon de pacientes con SII.
- Un estudio de 2020 encontró niveles plasmáticos elevados de la citocina antiinflamatoria IL-10 en pacientes SII (contradiciendo a los estudios anteriores), así como de la citocina proinflamatoria interferón gamma (INF-γ), que recibe este nombre por su capacidad de interferir en la replicación vírica, y que es producida principalmente por células NK (Natural Killer) y NKT (Natural Killer T) durante la respuesta inmune innata (o primaria) y por linfocitos T colaboradores CD4 y linfocitos T citotóxicos CD8 durante la respuesta inmune adaptativa (o secundaria, cuando se ha creado una inmunidad específica contra el antígeno invasor, 2007), así como por células linfoides innatas (2015).
La alteración en los niveles de citoquinas puede deberse a varios motivos. Uno de ellos es la degranulación de los mastocitos, de la que ahora hablaremos. Otras veces son producidas por linfocitos T regulatorios y colaboradores (T regulatory cells o Tregs, y T helper cells o Th, la IL-10 proviene de ellos principalmente), linfocitos colaboradores de tipo 2 (Th2), eosinófilos, macrófagos (producen IL-6 y el TNF), monocitos (IL-6), y linfocitos Th17 (IL-17) (2020).Una de las células mencionadas, los linfocitos T reguladores, son los encargados de combatir patógenos intracelulares, y se comunican con el sistema nervioso entérico para invertir la producción de citoquinas antiinflamatorias a proinflamatorias. Un mecanismo para ello es la producción de CRF (hormona liberadora de corticotropina) ante un estrés psicológico o inmunológico, que puede unirse a receptores específicos en los mastocitos y hacer que liberen citoquinas proinflamatorias (y otros mediadores). Todo esto es importante porque un metaanálisis de 2018 encontró que, en biopsias de colon de pacientes con SII, había niveles aumentados de linfocitos T regulatorios CD3+ y, algo menos, CD4+. Desde otra línea de investigación, un estudio piloto de 2017 (con muestra muy reducida) encontró que los linfocitos T de pacientes SII-D presentaban signos de agotamiento, respondían menos a los estímulos, segregaban menos mediadores y se dividían menos, un tipo de respuesta, según los autores, a menudo observada en las infecciones crónicas.
Un estudio de 2020 estudió las células «tuft» en pacientes con SII-D/E no postinfeccioso. Estas células son «centinelas» químicos inusuales que se pueden encontrar en el epitelio intestinal, y que suelen responder a las infecciones de helmintos segregando la citocina proinflamatoria IL-25. En biopsias colónicas de pacientes SII-D no postinfeccioso, la densidad de estas células parecía estar aumentada, así como la secreción de IL-25. Los autores concluyeron que, en pacientes SII-D (no en SII-E), otros factores distintos de la exposición a helmintos parecen provocar un aumento en la secreción de IL-25.
MASTOCITOS
Un metaanálisis de 2017, realizado sobre 22 estudios, encontró incrementos en los mastocitos y en los linfocitos T CD3+ en pacientes con SII frente a sujetos sanos, variando estos incrementos según la sección del colon que se biopsiara y el tipo de SII (diarrea VS estreñimiento). Estudios anteriores habían encontrado aumentos en el número de mastocitos en distintas secciones del intestino de pacientes con SII, incluyendo biopsias del recto (SII-D) , del sigma, del colon descendente, del colon ascendente (SII-D), ciego (SII-D), íleon terminal (SII-D), yeyuno (SII-D) y duodeno.
Estos mastocitos contienen gránulos con mediadores como la histamina, la triptasa, o los factores de crecimiento nervioso (NGF en inglés) entre otros, capaces de modular la sensibilidad visceral, la motilidad gastrointestinal, las secreciones epiteliales, la permeabilidad epitelial y la barrera mucosa, y la respuesta inmunitaria e inflamatoria intestinal (revisión de 2016). Los mastocitos liberan los gránulos con estos mediadores a través de un proceso llamado «degranulación». El número de mastocitos, no obstante, parece no ser tan importante en el SII como su nivel de actividad. En un estudio de 2004 se demostró que la presencia de mastocitos cerca de los nervios de la mucosa colónica, activos y liberando grandes cantidades de histamina, correlacionaba con la severidad y la frecuencia del dolor abdominal en pacientes con SII. Otro estudio, de 2019, reforzó la teoría antes mencionada de que el número y localización de los mastocitos no es relevante de cara a los síntomas, volviendo a poner el foco en su nivel de activación (la medida en que liberan mediadores como histamina, serotonina, o proteasas como la triptasa).
Los mastocitos y su rol en el SII llevan varias décadas siendo objeto de una investigación intensiva, a pesar de la cual, los resultados hasta el día de hoy han sido frustrantes, con pocas respuestas y un sinfín de nuevas preguntas. Las medicaciones utilizadas para tratar esta diana terapéutica, como los estabilizadores mastocitarios (cromoglicato disódico y ketotifeno) o antihistamínicos (ebastina o el propio ketotifeno) han demostrado una eficacia prácticamente nula en la práctica, pese a contar con algunos estudios aislados a su favor (ver el apartado 4 sobre controversias).
Para entender mejor el rol de los mastocitos en el SII, podemos guiarnos por el esquema realizado por Zhang en una revisión de 2016:
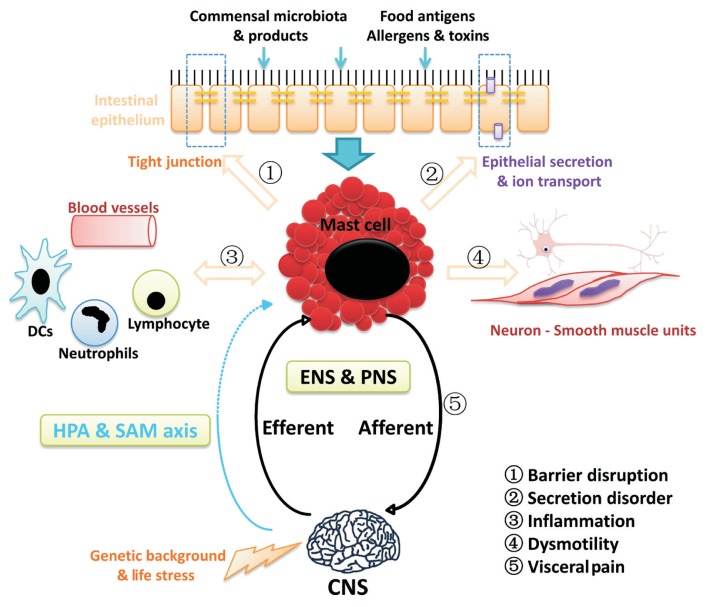
En esta primera imagen se representan las distintas vías por las que los mastocitos pueden influir sobre la fisiopatología del SII.
Lo primero es entenderlos en su contexto, y por esto se nos muestra a grandes rasgos el «ecosistema» que los rodea.
Arriba del todo, se nos representa la barrera epitelial, dejando por encima los contenidos del tubo digestivo (lumen) y justo por debajo el interior de las células epiteliales y tejidos internos del intestino. Podemos ver que en el tubo digestivo hay bacterias comensales (nuestra microbiota), y productos generados de la fermentación de estas bacterias (ver apartado de alteraciones de la microbiota para más información). También hay antígenos alimentarios, alérgenos y toxinas. Todos estos contenidos del lumen pueden influir directa/indirectamente en el nivel de actividad de los mastocitos.
Del mismo modo, el estrés psicológico y los acontecimientos vitales negativos contribuyen a la degranulación (=activación) mastocitaria a través de vías fisiológicas directas (la inervación nerviosa periférica) e indirectas (el eje hipotalámico-pituitario-adrenal/HPA o el eje simpático-adrenal-medular/SAM).
Una vez que los mastocitos se activan, podrían influir en los síntomas del SII de diversas maneras (seguir la numeración de la foto):
- Regulan la permeabilidad intestinal actuando sobre las proteínas de unión estrecha.
- Regulan el transporte epitelial de agua e iones.
- Regulan el flujo sanguíneo y las funciones endoteliales, así como la inmunomodulación, la inflamación, y la defensa contra los microbios.
- Regulan la peristalsis intestinal.
- Regulan las aferencias (señales al cerebro) de la sensación visceral, por mecanismos neuroinmunes.
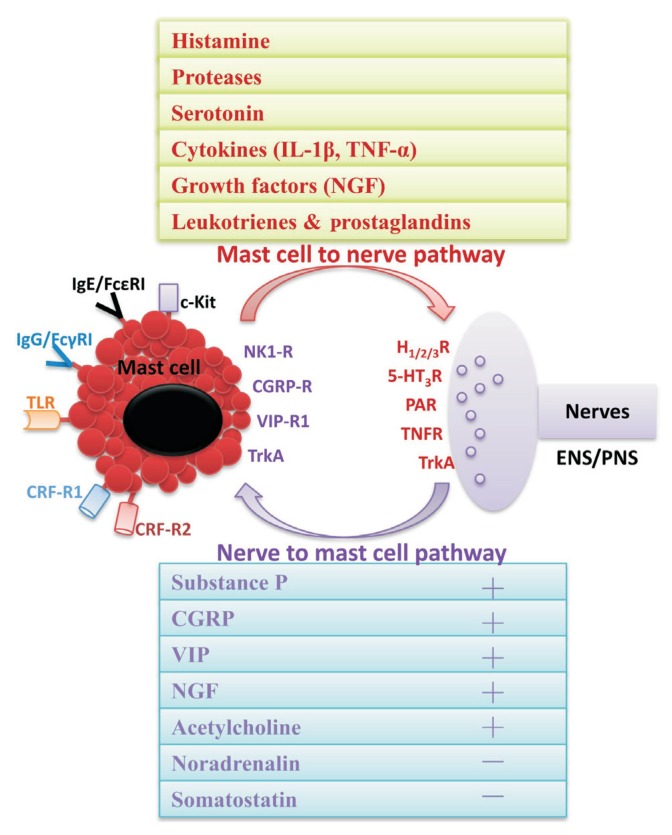
En esta otra imagen se nos muestra la relación bidireccional entre mastocitos y neuronas del sistema nervioso entérico.
Por un lado, el mastocito, al activarse, segrega sustancias bioactivas almacenadas en gránulos (histamina, serotonina, enzimas proteasas) o recién sintetizadas (citocinas, factores de crecimiento, metabolitos lipídicos) que actúan en determinados receptores de las terminaciones nerviosas, resultando principalmente en un aumento del dolor visceral. Estas sustancias tienen una función proinflamatoria, aunque llama la atención que la serotonina sea una de ellas… ¿cómo va a ser proinflamatoria la serotonina si los antidepresivos – que aumentan los niveles de serotonina – modulan y reducen el dolor? Esto se debe a que la serotonina actúa de maneras distintas según su localización. La serotonina presente en las mucosas (segregada sobre todo por las células enteroendocrinas de tipo enterocromafines) es PROinflamatoria, mientras la serotonina presente en las sinapsis neuronales (la que aumenta con el uso de neuromoduladores) es ANTIinflamatoria.
Por otro lado, cuando la neurona interactúa con el mastocito, tanto las neuronas intrínsecas (las propias del intestino, del sistema nervioso entérico, que se localizan en los plexos mientérico y submucoso descritos en el apartado de alteraciones neurológicas) como las extrínsecas (que llevan las señales del SNE al cerebro – aferencias – y del cerebro al SNE – eferencias -) pueden responder a toda una serie de estímulos mecánicos, biológicos y químicos, liberando una elevada cantidad de neuropéptidos, como la sustancia P (SP), el polipéptido intestinal vasoactivo (VIP), el péptido relacionado con el gen de la calcitonina (CGRP), el factor de crecimiento nervioso (NGF) y la somatostatina, todos los cuales regulan la actividad mastocitaria.
En el esquema también se representan otros elementos, como los receptores/mediadores mastocitarios:
- IL-1β: Interleucina – 1 beta (una citocina proinflamatoria)
- TNF-α: Factor de necrosis tumoral – alfa (otra citocina proinflamatoria). En experimentos in vitro con biopsias del íleon de cerdos, se ha mostrado que el TNF-alfa aumenta la permeabilidad (2012) de sus células epiteliales, al inducir una desregulación de las proteínas de unión estrecha.
- IgE/FcɛRI: Receptor de alta afinidad para la IgE (las alergias alimentarias por IgE están mediadas por los mastocitos)
- IgG/FcγRI: Receptor de alta afinidad para la IgG, inmunoglobulina sintetizada por el cuerpo en respuesta a la invasión por bacterias, hongos o virus
- TLR: Receptor tipo Toll. Estos receptores responden a ligandos llamados «patrones moleculares asociados a patógenos» (PAMPs, por su nombre en inglés) presentes en bacterias (sobre todo los lipopolisacáridos en la superficie de bacterias gramnegativas -las que se tiñen de color rosado en la tinción de Gram- ), hongos, virus, o en la propia célula, entre otros. La activación de estos receptores activa dos vías de señalización diferentes. En una de ellas, dependiente de la proteína MyD88 (que actúa como un adaptador, conectando proteínas que reciben señales desde fuera de la célula a las que transmiten señales dentro de ella), todos los receptores TLR (10 en esta revisión de 2012) excepto el 3, activan esta proteína, que activa a NF-kB, un complejo proteico que regula la transcripción del ADN celular, y que en este caso induce la producción de citocinas inflamatorias como IL-1, IL-8, TNF-alfa, e IL-12. Cuando se une a TLR-7 o TLR-9, MyD88 también puede inducir la producción de interferón de tipo 1 (IFN) , otra citocina proinflamatoria. En la otra vía de señalización, independiente de MyD88, los receptores TLR-3 y TLR-4 activan al adaptador TRIF (que tiene un rol parecido a MyD88), que a su vez activa a NF-kB por un mecanismo similar, y que además induce la producción adicional de IFN-β. Los mastocitos tienen receptores TLR 1-7 y 9, aunque parece que no se degranulan por la activación de éstos, o al menos eso dicen los estudios in vitro. En pacientes con SII, un metaanálisis de 2019, que repasó 8 estudios, encontró que la expresión de los TLR 2, 3, 4 y 5 estaba aumentada, mientras que la de los TLR 7 y 8 estaba disminuida. En las conclusiones, apuntaban a un posible rol de las bacterias y virus intestinales en las alteraciones de los TLR, y en la patofisiología del SII.
- CRF-R1 y CRF-R2: Receptores 1 y 2 del factor liberador de corticotropina. La corticotropina es un péptido hormonal que se produce en la respuesta de estrés, uniéndose a estos receptores y ayudando a la activación/degranulación de los mastocitos (todo parece indicar que esta sea una vía fisiológica por la que la ansiedad y el estrés influyen en la sintomatología del SII) . De hecho, en cerdos se ha demostrado que el CRF provoca un aumento de la permeabilidad paracelular mediante la liberación de proteasas y TNF-alfa en los mastocitos, algo que también ocurre en las EII (Crohn, colitis). Es improbable que esta respuesta al estrés esté mediada exclusivamente por los mastocitos, pues los receptores CRF-R1 y CRF-R2 también se encuentran en diversas células y tejidos del tracto gastrointestinal (2018), como las células enteroendocrinas de tipo enterocromafines, las células epiteliales (CRF-R1) y caliciformes, los macrófagos, o los plexos submucoso (CRF-R1) y mientérico.
- NK-1: Receptor 1 de neuroquinina, es el receptor al que se une la sustancia P, un polipéptido clave en la percepción del dolor.
- VIP-R1: Receptor 1 del polipéptido intestinal vasoactivo (VIP). El VIP es conocido por estimular la secreción de agua y electrolitos en el intestino, aumentando la motilidad, aunque también se presenta en otros órganos, desempeñando otras funciones
- TrkA: Receptor quinasa A tropomiosina, también llamado receptor de alta afinidad al factor de crecimiento nervioso (NGF, uno de los neuropéptidos proinflamatorios que la neurona envía al mastocito, degranulándolo, y que el mastocito también envía a las neuronas)
O los receptores/mediadores neuronales:
- HR-1, HR-2 y HR-3: Receptores de histamina 1, 2 y 3. Los antihistamínicos actúan sobre el receptor 1, reduciendo la respuesta alérgica
- 5-HT3R: Receptor 3 de la 5-hidroxitriptamina (serotonina). Es la diana terapéutica de tratamientos como el ondansetrón/alosetrón, antagonistas que «bloquean» este receptor, para inhibir la motilidad intestinal
- PAR: Receptor activado por proteasas (como la triptasa y quimasa segregadas de los gránulos del mastocito, y que se utilizan como marcadores de la activación de éstos en los estudios científicos). Hay 4 tipos (PAR1, PAR2, PAR3, PAR4) que se activan con proteasas como la trombina (1, 3 y 4) o la tripsina, triptasa, catepsina S o elastasa (2), entre otras. Los receptores PAR cumplen un amplio abanico de funciones, desde la hemostasia (que permite la coagulación/cicatrización) o la trombosis, ambas mediadas por la trombina, hasta la regulación del tono vascular y la permeabilidad vascular (que permite la inflamación y formación de edemas como mecanismo de defensa) clave en enfermedades como la arteriosclerosis o la restenosis, pasando por una evidencia creciente de su rol en el crecimiento y diferenciación muscular y óseo. Pero lo que a nosotros nos interesa es que los receptores PAR2 se han vinculado con la hiperactivación crónica de nociceptores en el SII, teniendo un más que probable rol en la sensación de dolor. Recientemente se han propuesto antagonistas del receptor PAR2 como un posible tratamiento para atacar esta diana terapéutica. El receptor PAR1 también se ha vinculado con el SII, comprobándose que su activación por diversas proteasas (como la elastasa 3a, quimotripsina C, la subunidad tipo beta-2 del proteosoma, o una isoforma inespecífica del complemento C3, entre otras) se traduce en activación nerviosa sólo en el SII, versus la colitis ulcerosa (donde el PAR1 no parece implicado en la activación nerviosa).
- TNFR: Receptor del factor de necrosis tumoral
- TrkA: Receptor quinasa A tropomiosina, también llamado receptor de alta afinidad al factor de crecimiento nervioso (NGF, uno de los neuropéptidos proinflamatorios que la neurona envía al mastocito, degranulándolo, y que el mastocito también envía a las neuronas)
La neurona a su vez se comunica con el Sistema Nervioso Entérico (ENS) , y éste con el Sistema Nervioso Periférico (PNS).
Para dar un poco de orden a toda esta información, Guy Boeckxstaens, digestivo belga experto en SII, y miembro del Translational Research Center for Gastrointestinal Disorders del Hospital Universitario de Leuven, comentó en una entrevista de 2018 que, hasta el momento, conocemos un mínimo de 3 vías por las que la activación mastocitaria (no su localización, ni su número) podría influir sobre la hipersensibilidad visceral del SII.
- La primera vía es una activación directa de las terminaciones nerviosas por la liberación de mediadores diversos, siendo los más conocidos las proteasas, la histamina y la serotonina (habría que añadir las citoquinas). Cuando las terminaciones nerviosas son activadas, envían señales de dolor al cerebro a través de la médula espinal.
- La segunda vía es la liberación de factores de crecimiento nervioso (NGF) que regularían al alza la expresión de moléculas nociceptivas en las terminaciones nerviosas. Algunas de estas moléculas, presentes en el SII y en diversos trastornos de dolor somático, serían los canales TRP (potencial de receptor transitorio), incluyendo el TRP vaniloide 1 (TRPV1), el TRP vaniloide 4 (TRPV4) o el TRP ankirina 1 (TRPA1). Si la expresión de estos receptores sensoriales del dolor está aumentada por los NGF, se produce hipersensibilidad visceral.
- Y la tercera vía es la sensibilización de estos mismos receptores TRP (TRPV1, TRPV4, TRPA1) por la liberación de mediadores mastocitarios como la histamina, haciendo que las terminaciones nerviosas respondan más intensamente a los estímulos.
Y con esto ya tenemos una «idea general» del rol de los mastocitos en el SII.
ÁCIDO ARAQUIDÓNICO, DERIVADOS EICOSANOIDES Y PAR2
Otros reguladores inflamatorios son los ácidos grasos poliinsaturados (PUFA, PolyUnsaturated Fatty Acids), concretamente el ácido araquidónico (AA), que aparece asociarse a un perfil proinflamatorio en diversos trastornos inmunitarios. El ácido araquidónico es el precursor de unas moléculas llamadas eicosanoides, que tienen funciones inmunomoduladoras, y entre las que destacan las prostaglandinas E2 y los leucotrienos B4 (las prostaglandinas y leucotrienos son también mediadores segregados por los mastocitos, ver imagen superior) .
En mujeres con SII se ha demostrado la existencia de niveles aumentados de ácido araquidónico(AA) en el plasma sanguíneo, a través de un estudio de 2010 que utilizó cromatografía de gases. Y un derivado del ácido araquidónico, el eicosanoide ácido 5-oxo-eicosatetraenoico, aparece aumentado en subgrupos de pacientes con SII-E (estudio de 2018), donde podría inducir la hiperalgesia somática y visceral a través de un mecanismo no inflamatorio y mediado por el «receptor D acoplado a proteínas G relacionadas con MAS» (MAS-related G protein-coupled receptor D). Este receptor ya ha sido vinculado con la modulación del dolor, de hecho, los «receptores acoplados a proteínas G relacionadas con MAS» en general, están relacionados con el desarrollo, regulación y funcionamiento de las neuronas nociceptivas.
En un estudio de 2015, que utlizaba biopsias de colon, se comprobó que los pacientes con SII tenían niveles más altos que los controles de ácidos epoxieicosatrienoicos 5,6 (5,6-EET), un eicosanoide agonista de los receptores TRPV-4 (receptores que abren un canal iónico por el que pasan cationes Ca2+ y que están implicados en la sensación de dolor) y estos niveles correlacionaban con las puntuaciones de dolor e hinchazón. También se vio que la inducción de hipersensibilidad visceral en ratones , por sobrenadantes de biopsias de colon de pacientes con SII, era inhibida cuando se limitaba la expresión genética de los receptores TRPV-4 en estos ratones. Al final del estudio, se extrajeron metabolitos de los PUFA (eicosanoides y otros) de biopsias de pacientes con SII-D o de ratones con hipersensibilidad visceral inducida, y estos metabolitos activaron neuronas sensoriales de ratones en un modelo in vitro, a través de la activación del receptor TRPV-4. También se observó que, tras exponer neuronas sensoriales de ratones (in vitro) a sobrenadantes de biopsias de pacientes SII-D, éstas producían 5,6-EET a través de un mecanismo que implicaba al PAR2 y a la enzima citocromo epoxigenasa (CYPe). En este mecanismo, el 5,6-EET es metabolizado por la CYPe, produciéndose ácido araquidónico (AA), que se une a TRPV-4, activándolo. El receptor TRPV-4, al activarse, abre los canales de calcio (Ca2+) bajo su dominio, permitiendo la entrada de calcio extracelular y desencadenando la activación neuronal (por la carga iónica del calcio, que provoca un «potencial de acción» o impulso nervioso al cambiar la carga eléctrica de la membrana celular) y la hipersensibilidad. De hecho, en el estudio se comprobó que la inhibición de la citocromo epoxigenasa o de la SRC quinasa (SRC kinase) reducía la producción de 5,6-EET (y por tanto, la activación de TRPV-4) inducida por el PAR2. Esto confirma a la SRC quinasa y la CYPe como 2 mecanismos que vinculan el PAR2 a la activación de TRPV4 en las neuronas sensoriales del colon, al menos en ratones con sobrenadantes de SII-D.
Estudios anteriores (2008), también en ratones, ya habían relacionado los receptores PAR2 y TRPV-4, aunque está por ver si este efecto se repite en humanos. Abajo, una foto resumen:
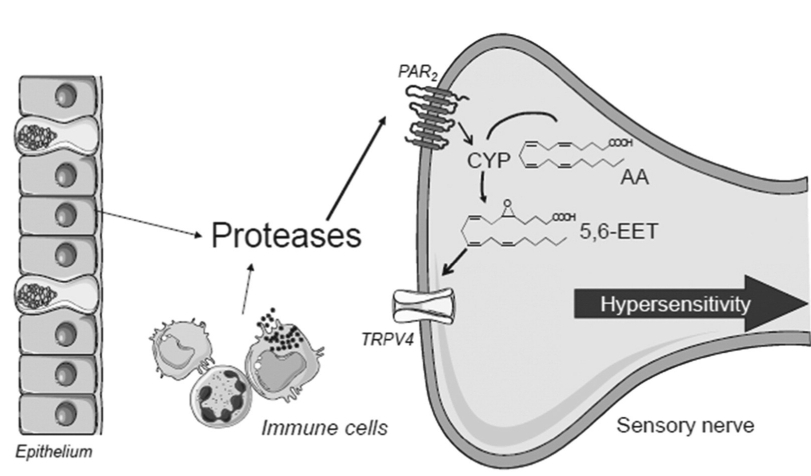
Un año antes (2007), un estudio mostró que en biopsias de pacientes con SII había una actividad proteolítica (escisión del aminoácido arginina) 2-3 veces superior a la normal en SII (6 veces en las EII), y que era dependiente de NF-kB (factor implicado en la transcripción del ADN y diversos mecanismos inmunitarios), pues al incubar las biopsias de SII con un inhibidor de NF-kB (BAY 11-7085) la actividad proteolítica se reducía. Esto se confirmó con un estudio in vivo, utilizando «lavados» de colon (se inyectaba una solución salina en el recto de pacientes SII y controles, se extraía a los 30 segundos y se congelaba), donde la actividad proteolítica de los SII era 5 veces mayor. Las biopsias de SII también expresaban y liberaban niveles más altos de enzimas como tripsina y triptasa. Sin embargo, lo más interesante es que, cuando cultivaron sobrenadantes de biopsias de SII con neuronas sensoriales de ratones, éstas se sensibilizaron, sensibilización que podía prevenirse con un inhibidor de serina proteasas (las serina proteasas son un tipo de enzimas entre las que se incluye la tripsina), y que directamente noocurría en neuronas cuyo PAR2 (receptor activado por proteasas tipo 2) noerafuncional. Cuando estos sobrenadantes se administraban en el colon de ratones, les provocaban hiperalgesia visceral y alodinia, pero, nuevamente, este efecto podía bloquearse con inhibidores de serina proteasas y un antagonista PAR2, y si los ratones no tenían receptores PAR2, la hiperalgesia visceral y la alodinia eran inexistentes. En conclusión, el estudio mostró que la activación de PAR2 parece ser clave en la generación de los síntomas de hipersensibilidad visceral del SII, como mínimo en modelos animales.
En relación esto, un estudio de 2017 encontró que, en pacientes con SII, el epitelio intestinal produce y libera la proteasa activa tripsina-3, que puede señalizar a las neuronas entéricas y provocar hipersensibilidad visceral en un proceso dependiente de los receptores PAR2. La tripsina-3 parecía la única de las 3 formas estudiadas de esta enzima que estaba sobreexpresada en células epiteliales estimuladas y en tejidos de pacientes con SII. Esta proteasa era capaz de señalizar a neuronas entéricas submucosas humanas y a neuronas sensoriales de ratones, así como de provocar hipersensibilidad visceral invivo a través del PAR2.
ANOTACIONES FINALES
Todos estos estudios se han centrado mayoritariamente en analizar estos procesos a nivel del colon, que parece ser el órgano más afectado en el SII (en el sentido de tener un funcionamiento deficiente) . Sin embargo, parece que el intestino delgado también podría presentar algunas de las alteraciones que hemos comentado en este apartado. En un estudio de 2006 de la revista «Gut», se encontraron incrementos leves en los linfocitos intraepitaliales CD3+, y más notables en la actividad mastocitaria, en el yeyuno de pacientes con SII-D. En otro estudio, de 2012, destacado en «Nature», encontraron un posible aumento en la permeabilidad de dicho área, asociado al aumento de la actividad mastocitaria, y asociadas ambas alteraciones a la sintomatología de los pacientes, aunque la muestra fue muy pequeña, y la evidencia es muy preliminar. Otro artículo, de 2016, publicado en el Journal of Gastroenterology and Hepatology, realizó una breve revisión sobre las alteraciones de la barrera intestinal en el SII, alteraciones que, según los investigadores, aparecen en ambos tramos del intestino, delgado y grueso, y en todos los subtipos de SII. Probablemente el tiempo, y las mejoras tecnológicas, podrán esclarecer mejor el rol del ID en estos problemas.
Estos fenómenos (permeabilidad, microinflamación, alteraciones inmunitarias) concentran gran parte de la investigación actual en SII, pero no están exentos de polémica. Las técnicas actuales para medir la permeabilidad intestinal son muy susceptibles de mejora, por lo que es difícil sacar conclusiones sólidas de las investigaciones en este terreno. No sólo el SII, sino muchas otras enfermedades presentan alteraciones de la permeabilidad, y sigue sin esclarecerse si esta permeabilidad es una causa de los síntomas, o simplemente un efecto colateral de la hiperactividad del sistema inmune.